

Estudo traz evidências de que vírus SARS CoV-2 surgiu a partir dos processos naturais de evolução dos seres vivos.
Por: Júlio Bernardes
Fonte: Editorias de Atualidades, Ciências, Ciências Biológicas do jornal.usp.br/?p=308206
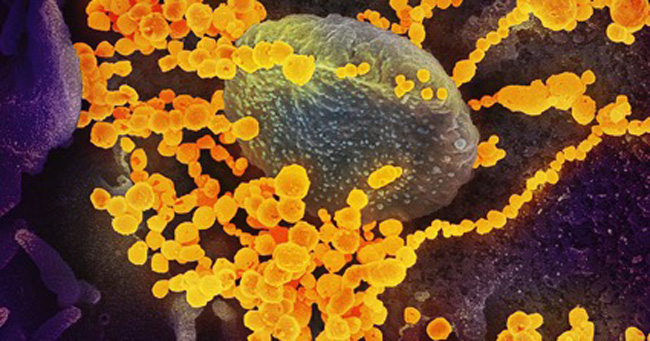
Mutações do genoma do vírus que o tornam mais infeccioso em humanos surgem aleatoriamente durante sua replicação e, por serem imperfeitas, é pouco provável a hipótese de terem sido produzidas pelo homem – Foto: NIAID via Wikimedia Commons (CC 2.0)
Quando a epidemia de covid-19 começou a avançar pelo mundo, surgiu uma grande controvérsia sobre a origem do vírus SARS CoV-2, que provoca a doença. Houve até quem dissesse que o vírus foi manipulado ou mesmo fabricado em laboratório. No entanto, um estudo de pesquisadores dos Estados Unidos, Escócia e Austrália, descrito em carta publicada na revista Nature Medicine, em 17 de março, traz evidências de que o SARS CoV-2 surgiu a partir dos processos naturais de evolução dos seres vivos.
O texto aponta mutações no genoma do vírus que o tornam mais infeccioso em seres humanos e que surgem aleatoriamente durante sua replicação. Essas mudanças são imperfeitas, o que torna improvável a hipótese de terem sido produzidas pelo homem.
O Corona vírus
“Os vírus têm genomas que não são muito grandes, então é possível sequenciá-los por inteiro de maneira bastante confiável, e estabelecer comparações entre as diversas sequências”, comenta o professor Daniel Lahr, do Departamento de Zoologia do Instituto de Biociências (IB) da USP. Ele não participou do estudo, mas comentou o artigo a pedido do Jornal da USP. “O vírus SARS CoV-2, causador da covid-19, tem um genoma com cerca de 30 mil bases, enquanto o genoma humano tem aproximadamente 3 bilhões de pares de bases e a bactéria Escherichia coli, cujo uso é muito comum em experimentos laboratoriais, tem de 4 a 5 milhões de pares de bases.” Bases são as unidades moleculares que formam o DNA.

Em Wuhan, na China, onde surgiram os primeiros casos da Covid-19, populares comemoram o fim do distanciamento social.
Para fazer as comparações, o professor conta que há uma série de fórmulas matemáticas que determinam como as sequências do genoma estão relacionadas. “Todas as sequências são colocadas em uma grande matriz para comparar as mutações e substituições do genoma”, explica. “Como já existem dados genéticos sobre a evolução de uma grande quantidade de organismos, as árvores filogenéticas, há modelos teóricos que explicam como esses processos devem ocorrer e permitem fazer uma série de predições sobre o que aconteceu durante a história evolutiva do vírus.”
De acordo com Lahr, os pesquisadores, ao analisarem as variações de todo o genoma do vírus, conseguiram determinar que o SARS CoV-2 é muito proximamente relacionado com um vírus já descrito em morcegos, o RATG13. “Isso significa que eles possuem um hipotético ancestral comum”, destaca, “porém a observação de partes específicas do genoma indica semelhanças que, na comparação com outros vírus, dão a oportunidade de explicar eventos de evolução e identificar as mutações mais importantes para infeccionar seres humanos”.

A análise do conjunto do genoma do SARS CoV-2 indica que ele é muito proximamente relacionado com o vírus RATG13, já descrito em morcegos, indicando a existência de um hipotético ancestral comum; parte do genoma relacionada com infecção em humanos se assemelha à do vírus que atinge o pangolim (exemplar na foto), mamífero aparentado com o tatu, embora seja preciso analisar mais amostras recolhidas de animais para determinar origem do SARS CoV-2 -Foto: Reprodução/Nature Medicine
Mutações
Segundo o professor do IB, as mutações acontecem de forma aleatória, durante a replicação dos vírus no interior das células. “Embora a taxa de erro seja muito pequena, são replicados milhares de genomas virais ao mesmo tempo, ao longo de vários dias, dando origem a modificações aleatórias”, relata. “A maioria dessas mudanças são inviáveis para o vírus, mas uma pequena parte delas irá ser potencialmente adaptativa, fazendo com que o vírus consiga infectar um novo hospedeiro e amplie sua área de atuação.”
Sabendo que os genomas acumulam mutações, os cientistas conseguiram encontrar evidências de mudanças que não deram certo e outras que podem ter ajudado a infectar os seres humanos de forma mais eficiente. “Todos esses indícios permitiram deduzir que o padrão geral de mutações do SARS CoV-2 corresponde aos modelos evolutivos existentes”, afirma Lahr. “Assim, os pesquisadores apontam duas hipóteses para a ocorrência dessas mutações: uma, que teriam acontecido ainda no reservatório animal do vírus, que ainda não é conhecido, e outra, que a diversificação teria acontecido após a invasão nos seres humanos. Todos esses indícios permitiram deduzir que o padrão geral de mutações do SARS CoV-2 corresponde aos modelos evolutivos existentes.”

Teste de DNA
O professor relata que o estudo identificou uma mutação no genoma do SARS CoV-2 relacionada com a produção da proteína SPIKE, que ajuda o vírus a aderir à superfície das células e introduzir-se nelas para se replicar. “Em seres humanos, a proteína se liga a um receptor conhecido como ACE2, presente nas células do pulmão, que são invadidas de forma muito intensa, provocando a doença”, descreve. “Como essa ligação, apesar de eficiente, não é perfeita, é pouco provável que a mutação tenha sido produzida em laboratório.”
Embora a mutação descrita no trabalho seja parecida com a encontrada no genoma de vírus presentes no pangolim (um mamífero carnívoro da ordem Pholidota), Lahr aponta que é preciso analisar um maior número de amostras recolhidas de animais para determinar com precisão a origem do SARS CoV-2. “A publicação demonstra a importância de entender a trajetória evolutiva do vírus para, juntamente com estudos bioquímicos sobre sua introdução nas células, criar futuras estratégias de prevenção e tratamento da covid-19”, destaca. “As vacinas contra a gripe, por exemplo, são produzidas anualmente a partir da coleta de amostras e da identificação das variedades de vírus mais infecciosas.”
Mais informações: e-mail dlahr@ib.usp.br, com Daniel Lahr